Query Allele Frequency
Filter variants by population allele frequency using gnomAD data, the largest database of human genetic variation. Identify rare, common, or population-specific variants for your research.
The Genome Aggregation Database (gnomAD) is the largest collection of human genetic variation data, containing allele frequencies from diverse global populations.
Learn more in the gnomAD overview.
Quick Filtering with MAF
The simplest way to filter by rarity is using minor allele frequency (MAF). This searches across both gnomAD genomes and exomes datasets simultaneously for comprehensive population frequency filtering.
Common MAF Thresholds
maf < 0.001maf < 0.01maf < 0.05Example: Finding Rare Variants
Find rare variants (frequency < 1%) using maf < 0.01:
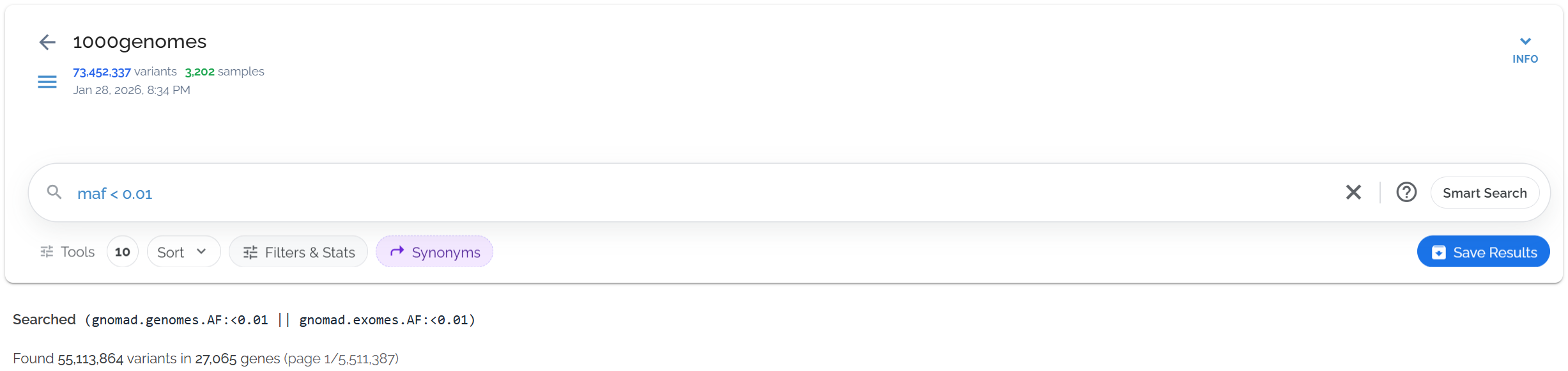
Using MAF to quickly identify rare variants across all gnomAD populations
maf as a query automatically searches both gnomAD genomes and gnomAD exomes datasets, providing comprehensive frequency filtering without needing to specify individual datasets.Population-Specific Searches
For more precise population genetics analysis, query specific gnomAD population datasets using exact field names. This lets you identify variants that are rare in one population but common in another.
Example: Non-Finnish European Population
Search for variants rare in the Non-Finnish European population using gnomAD.genomes.af_nfe < 0.01:
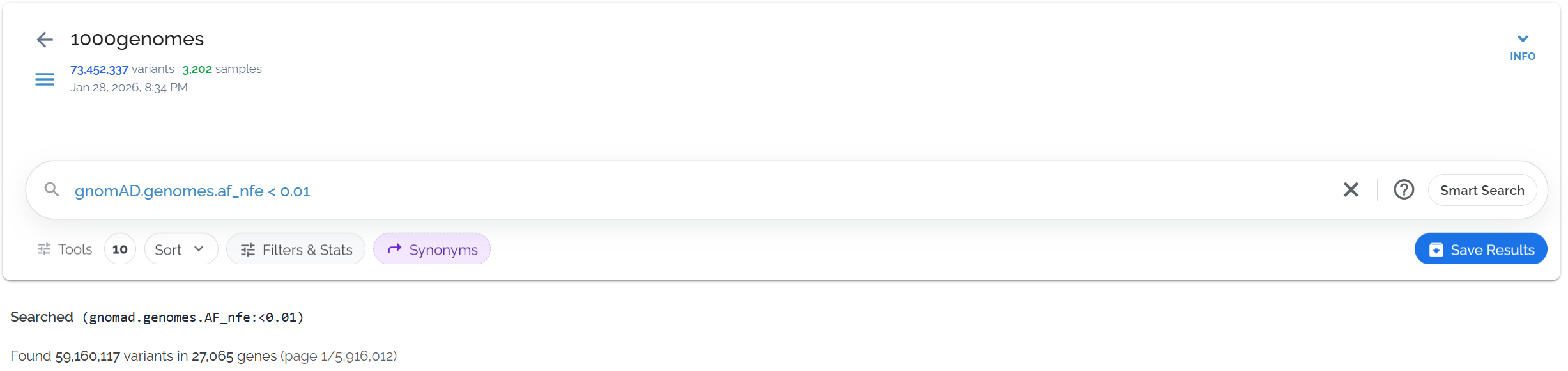
Filtering for variants rare in the Non-Finnish European population using specific gnomAD field names
Available gnomAD Populations
gnomAD Genomes
gnomAD.genomes.afAll populationsgnomAD.genomes.af_afrAfrican / African AmericangnomAD.genomes.af_amrLatino / Admixed AmericangnomAD.genomes.af_asjAshkenazi JewishgnomAD.genomes.af_easEast AsiangnomAD.genomes.af_finFinnishgnomAD.genomes.af_nfeNon-Finnish EuropeangnomAD.genomes.af_othOther
gnomAD Exomes
gnomAD.exomes.afAll populationsgnomAD.exomes.af_afrAfrican / African AmericangnomAD.exomes.af_amrLatino / Admixed AmericangnomAD.exomes.af_asjAshkenazi JewishgnomAD.exomes.af_easEast AsiangnomAD.exomes.af_finFinnishgnomAD.exomes.af_nfeNon-Finnish EuropeangnomAD.exomes.af_sasSouth Asian
Important Considerations
Allele Frequency Reporting
Bystro reports allele frequencies relative to the specific variant allele in your dataset, not all previously observed variants at that position (which is how dbSNP reports frequencies).
gnomAD IDs
gnomAD ID shows only one rs-number per variant. Learn how this can be used to create Set IDs for SKAT analysis in our FAQ section.
Practical Applications
Rare Disease Research
Combine ultra-rare frequency filtering with high CADD scores to identify potentially pathogenic variants:
maf < 0.001 AND cadd > 20Population Genetics
Compare allele frequencies between populations to identify population-specific variants:
gnomAD.genomes.af_eas > 0.05 AND gnomAD.genomes.af_nfe < 0.01Clinical Variant Filtering
Focus on clinically relevant rare variants in coding regions:
maf < 0.01 AND refSeq.exonicAlleleFunction:nonSynonymousConsider your study population: Use population-specific frequencies when studying specific ethnic groups.
Account for sample size: Check allele count (AC) and allele number (AN) fields for reliability.
Combine datasets: Use both exomes and genomes data for comprehensive frequency assessment.
Validate rare variants: Always validate ultra-rare variants with additional evidence.