Filter and Sort
Use Bystro's filtering tools and sorting options to narrow down your variants and organize them by the criteria most important to your research. Combine multiple filters with hierarchical sorting for precise variant selection.
Before diving into filtering, it's helpful to understand what each annotation field means. Our comprehensive field reference explains every available annotation.
View Complete Field Descriptions →Use the filter toolbar for precise control
The filter toolbar helps you filter by individual annotation fields while showing result counts for each option. Use checkmarks (✓) to include specific values and minus signs (-) to exclude them.
Let's filter to show only SNPs by excluding other variant types:

Open the 'type' filter and use minus boxes to exclude unwanted variant types
Filter by functional impact
Examine mutation types using the refSeq.exonicAlleleFunction field. This shows the coding impact of variants on protein sequences.
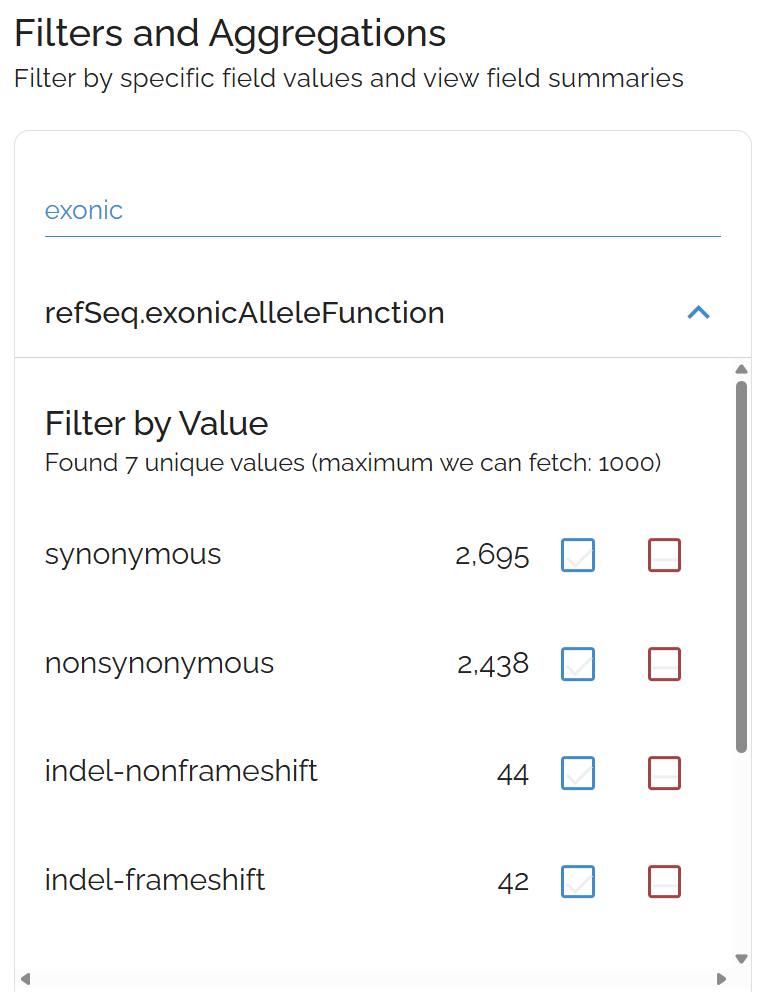
Use the exonicAlleleFunction filter to focus on specific mutation types like nonsense or missense variants
Common filtering strategies:
- Exclude
synonymousmutations to focus on protein-changing variants - Include only
stopGainfor nonsense mutations - Filter for
nonSynonymousmissense variants
Sort your filtered results
After filtering, use the Sort button to organize your results by genomic position, functional impact, allele frequency, or quality scores.
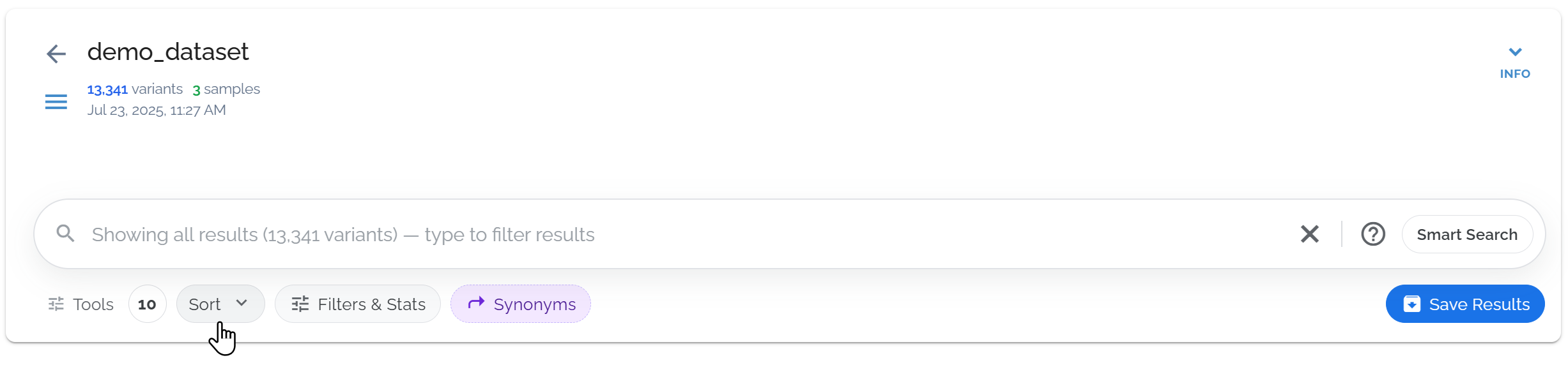
Use the sort button to sort by any sortable field
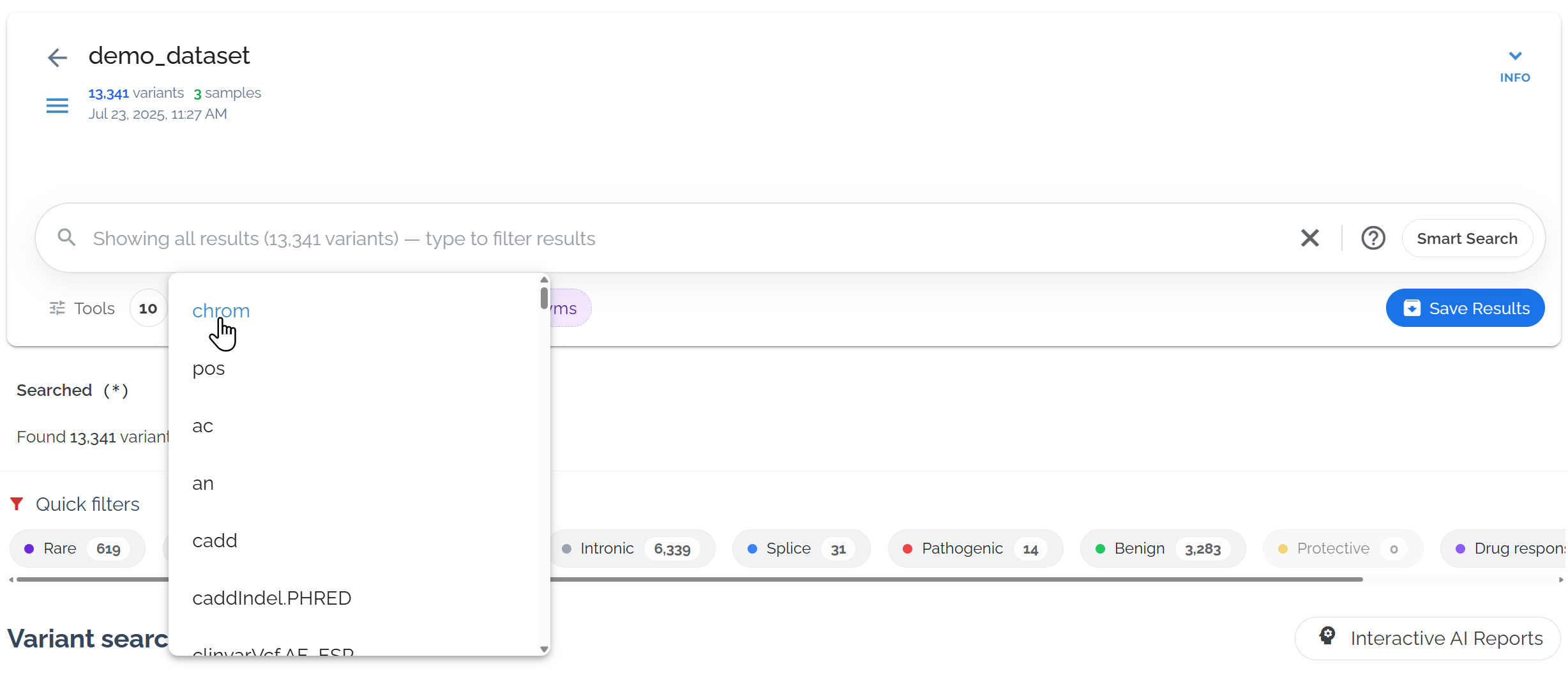
Sort by chromosome to organize variants by genomic location
- • First click: Sort by descending order (highest values first)
- • Second click: Sort by ascending order (lowest values first)
- • Third click: Remove that sort filter
- • Multiple sorts: Can be applied at the same time
Review your applied filters and sorts
Check the applied filters and sorts below the search bar. Click the gray bubble to see the exact query syntax that generates your results.
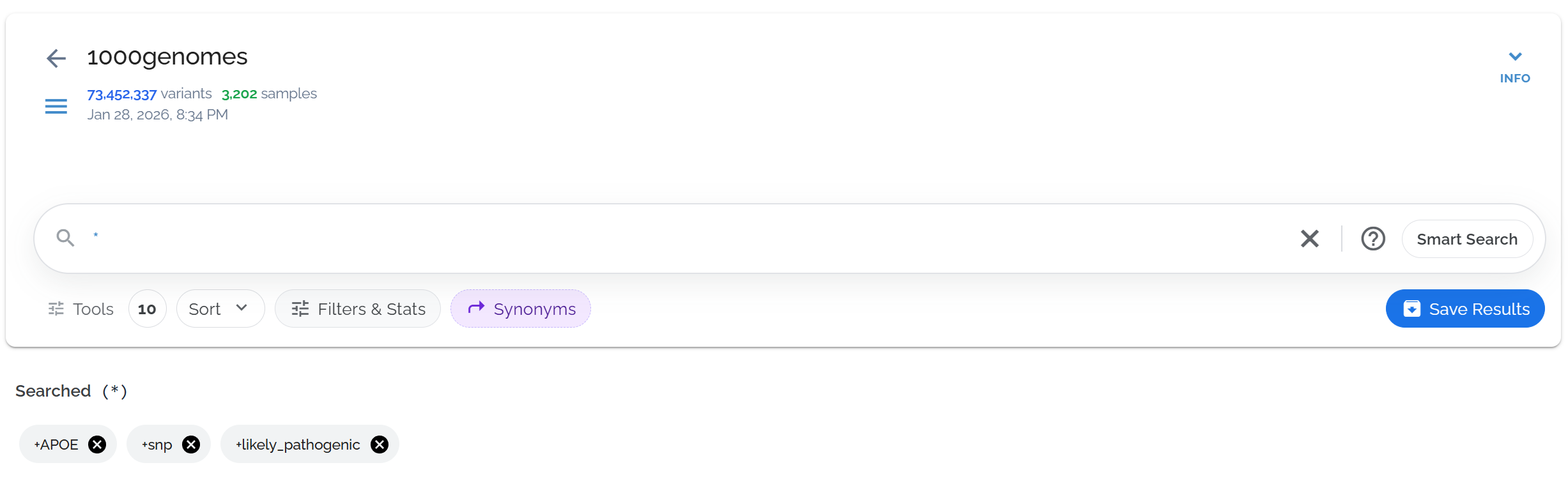
The gray bubble shows the selections from the filters toolbar, which can be clicked to reveal the underlying query
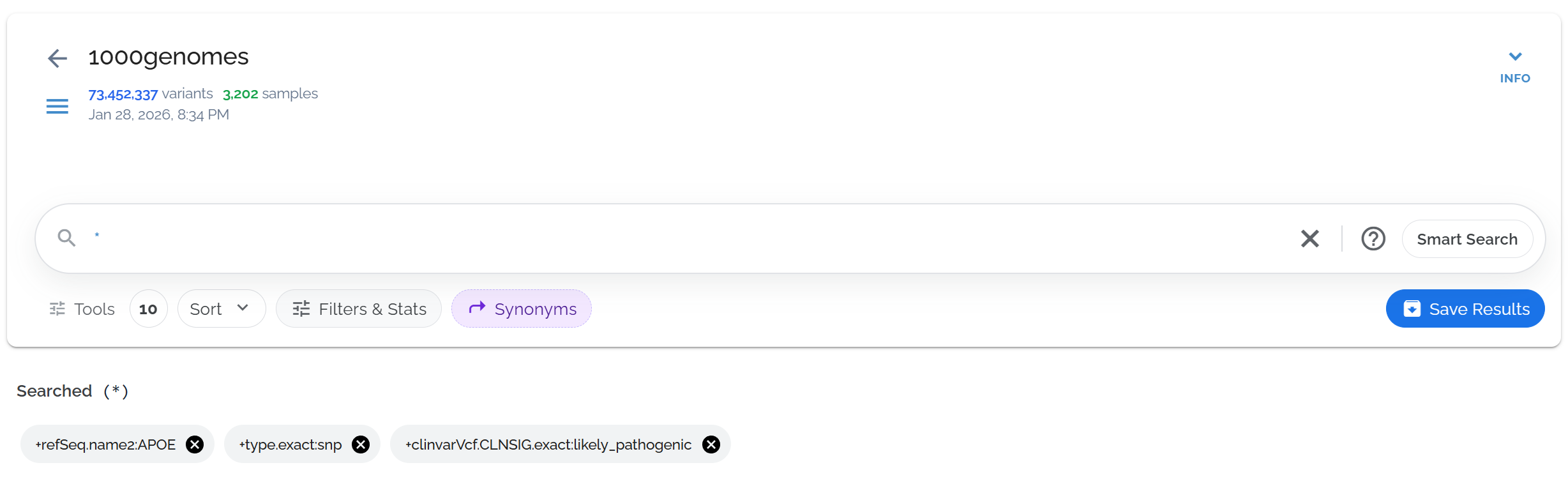
The gray bubble shows the underlying query syntax, useful for learning advanced search
Common Filtering Patterns
By Variant Type:
- •
type:SNP- Single nucleotide variants - •
type:DEL- Deletions - •
type:INS- Insertions
By Functional Impact:
- •
nonSynonymous- Protein-changing - •
stopGain- Nonsense mutations - •
synonymous- Silent mutations
Sorting Strategies by Research Goal
Research Priorities
High Impact Variants
Sort by CADD score (descending) to see potentially damaging variants first
Rare Variants
Sort by allele frequency (ascending) to identify private or ultra-rare variants
Genomic Regions
Sort by chromosome and position to browse systematically by location
Quality Control
Coverage Issues
Sort by read depth to identify poorly covered variants
Quality Scores
Sort by variant quality to review questionable calls
Population Outliers
Sort by allele frequency to spot potential artifacts
Advanced Filter Toolbar Examples
Sort by CADD Score
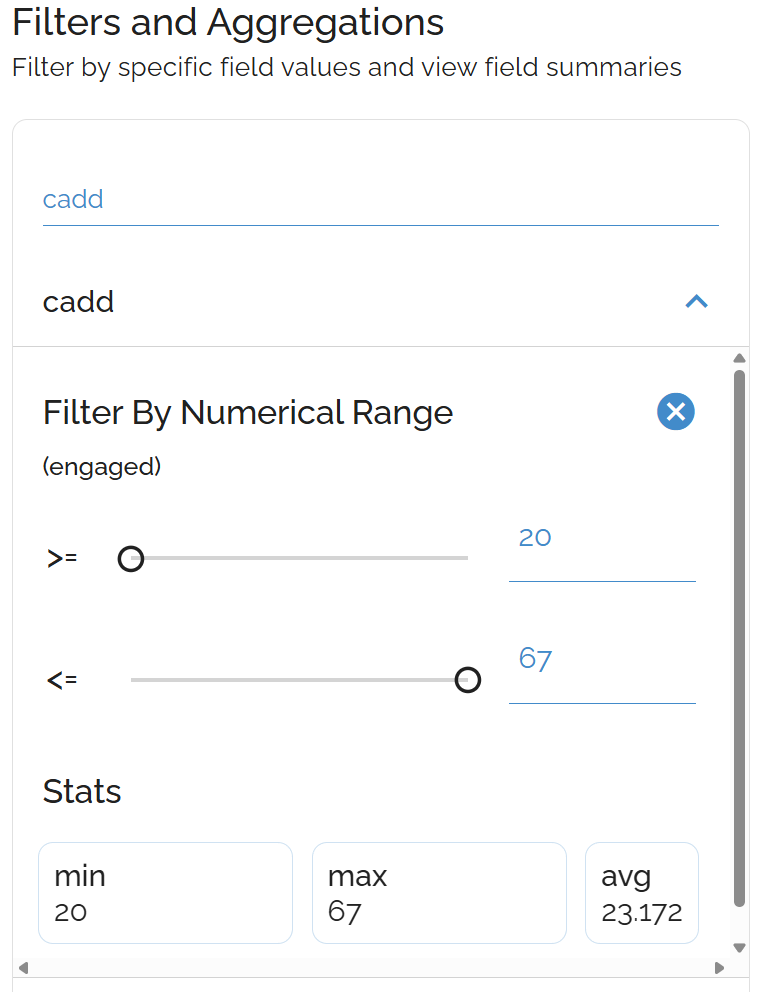
CADD scores predict variant deleteriousness
Sort by Allele Frequency
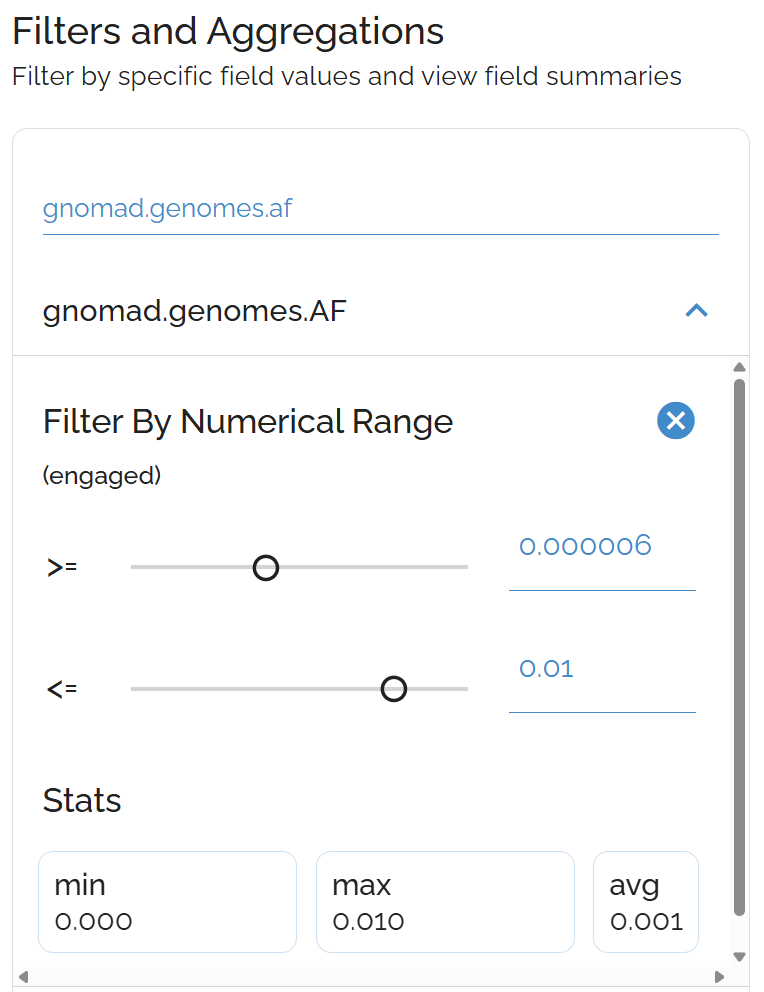
Identify rare variants by population frequency
Common Filter + Sort Combinations
Candidate Gene Analysis
- • Filter:
refSeq.name2:BRCA1+ Sort:cadd.phred(descending) - • Focus on high-impact variants within specific genes of interest
Clinical Prioritization
- • Filter:
clinvar.clinicalSignificance:Pathogenic+ Sort:gnomad.genomes.af(ascending) - • Prioritize pathogenic variants by rarity in the population
Systematic Analysis
- • Filter:
type:SNP AND nonSynonymous+ Sort:chrom+pos - • Systematic genomic analysis of protein-changing SNPs
• Filter first, then sort: Reduces the number of variants to organize
• Multiple sorts work hierarchically: Primary sort applied first, then secondary
• Indexed fields perform best: Common annotation fields are optimized
• Save useful combinations: Create saved searches for frequently used filter/sort patterns
Dataset used in examples: 1000 Genomes Project (73,452,337 variants in 27,192 genes, queries typically complete in ~0.5 seconds)